
Appendices
Appendices to ELSIE Risk Management of Leachables
Screening and Materials Selection
Appendix 1. HAP Hazard Grids
Figure 5
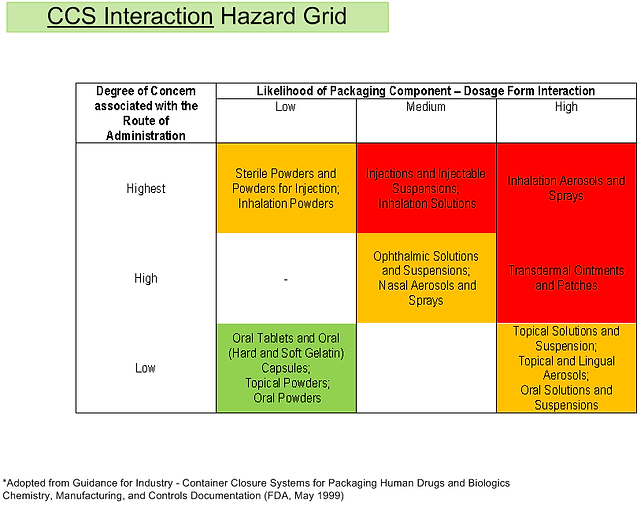
Figure 6

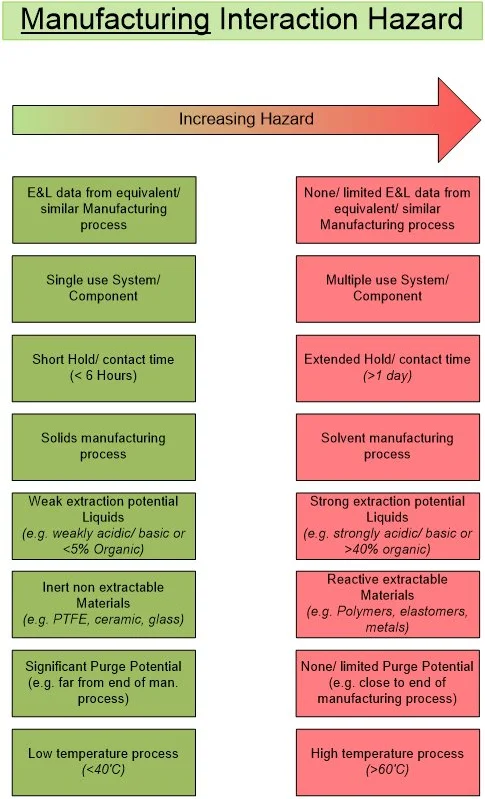
Figure 7
Appendix 2. Data and Compliance References Relevant to an E&L HAP
Technical Risk Assessment
Appendix 1. Additional information on knowledge impacting severity and probability scoring
This section provides a more detailed discussion of types of information on the materials a system is made of including their constituents with a potential to leach. This information is allocated to either the dimension of severity of harm or probability of occurrence. Information types are listed in descending order with respect to their significance in the risk assessment process.
A1.1 Severity of harm (severity)
Extractables data information
An extractable study is typically conducted with the intent to gain a complete understanding of what substances are present within a material and provide chemical characterization. Extraction solvents, as well as time, temperature and methodologies are chosen in such a way that materials of construction are subjected to more aggressive conditions in comparison to the environment in which the material is exposed to during manufacture, storage or delivery. These aggressive conditions ensure a broad range of chemical classes are extracted from the material at levels as close to the amount in the material as possible. Extractable data are toxicologically assessed and if no concerns in terms of patient safety are identified, the risk severity is anticipated to be decreased.
Biocompatibility information
Materials intended for use within a medical or pharmaceutical application should not exhibit biological reactivity, and as a minimum should comply with the requirements of USP<87>. More extensive in vivo biocompatibility testing (i.e., USP<88> and ISO10993) provides a greater degree of understanding of a material’s biocompatibility. Compliance with relevant tests in USP<88> (e.g., systematic injection and intracutaneous tests) and ISO10993 (e.g., parts 3, 5, 10 and 11) standards provide further reassurance that the material is appropriate for its application since an extract of it has not elicited a biological response in vivo. Where a material elicits an adverse biological response, material characterization would be necessary to identify the substance (and its source) that is causing this response in the animal. The extraction conditions and solvent systems employed in these studies are less aggressive than those applied during an extraction study. While it could be considered a more representative test for ascertaining the toxicological risk associated with leachables originating from container closure systems and manufacturing materials or components exposed to aqueous or lipid-based biopharmaceuticals, it is unable to characterize material(s), informing the amounts and identity of each substance within a material. Nevertheless, biocompatibility information related to a specific material or component can be used, if available, during the severity risk analysis as materials that have fulfilled compendial testing criteria (e.g., USP <87>, USP <88>, USP <661.2>) are deemed less severe in terms of risk.
Note: One can review the extraction conditions used for the biocompatibility test(s) to ascertain if they are applicable and relevant to the specific material and drug product being assessed. For example, biocompatibility data on a plastic that contacts an aqueous based (pH7) drug product formulation for short periods of time during manufacture could be used to understand the toxicological risk to the patient. However, it would be inappropriate to use biocompatibility data on its own to inform the toxicological risk associated with materials that are subjected to a highly organic environment for extended periods of time (e.g., MDIs).
Compendial compliance
Regulatory standards for plastic materials that are intended for use in pharmaceutical applications should be applied whenever possible. At a minimum, materials used within a pharmaceutical environment should meet these requirements and documentation to that effect should be available from the raw material manufacturer. Suppliers that modify the raw material into a component/product should be aware of these regulatory expectations and have gone through due diligence with their raw material supply chain, if the intent is to market the material for use in pharmaceutical and biopharmaceutical applications.
Medical grade plastics as defined by VDI (VDI 2017:2019-07, Medical Grade Plastics)[19] that have been demonstrated to be compliant with relevant monographs and regulations have a lower risk severity than materials not designated for the same purpose. Examples of compendial compliance include, but are not limited to, USP <661.1>, USP <661.2>, USP <381>, USP <660>, USP <381>, Ph.Eur. 3.1., Ph.Eur. 3.2.
Note: Additional testing by the end user might be required to complete the testing requirements. Consider the value and relevance of any missing data or information in light of the overall risk control strategy.
Non-compendial compliance
Information pertaining to elemental impurities levels can be used to evaluate the severity of the risk based on limits defined in ICH Q3D.
Information concerning food contact materials can also be leveraged to assess risk severity based on COMMISION REGULATION (EU) No 10/2011 of 14th January 2011 on plastic materials and articles intended to come into contact with food and the Code of Federal Regulations (CFR): 21CFR Parts 172 – 179, among others. At a minimum, materials used within a pharmaceutical environment should meet requirements described in these regulations for materials that are intended for use in food packaging applications. Whenever possible and applicable, compliance statements from the supplier should be procured. Suppliers that modify the raw material into a product should be aware of these regulations and should perform this due diligence if intent is to market material as suited to pharmaceutical and biopharmaceutical applications. Suppliers should also have systems in place to manage changes and communicate them to end users.
Compositional information
A detailed description of the monomers, additives package (e.g., antioxidants, nucleating agents, cure system, UV inhibitors, colorants, and etc.) and any processing aids (e.g., mold release agents) used to formulate and manufacture the material should be gathered whenever possible.
This information can be used to consider if chemicals used to manufacture the material present negligible risk to patient safety and have been used at the level previously endorsed by the regulatory agencies and are not on the Substances of Very High Concern (SVHCs) list within the Registration, Evaluation, Authorization, and Restriction of Chemicals (REACH) regulation within the EU.
Note: Substances included in REACH annexes are not to be considered a comprehensive list of extractables as degradation products and non-intentionally added substances are included. This list informs on what chemical entities are added to the material at the raw material supplier level.
Supplier information
Absence declarations for substances with high toxicological concern as natural latex, phthalates, nitrosamines, 2-mercaptobenzothiazole, and etc., should be procured, whenever available as it provides a means to decrease the severity associated with the identified risk.
A1.2 Likelihood of occurrence (probability)
Various aspects should be taken into consideration when assessing the probability of a risk to occur. The probability of the risk defines the likelihood of the risk event occurring to an extent that it would result in an adverse toxicological or product quality event. The probability of substances leaching during the manufacture, storage and/or administration of a biopharmaceutical product is complex and is best understood by evaluation of leachables data.
Leachables information
An analytical study that uses a variety of analytical methodologies which identify and demonstrate the absence of a broad range of chemical substances that have migrated into a commercially representative drug product formulation (subjected to storage at its normal or accelerated temperature and humidity environments over its shelf life) from materials used to manufacture, package and/or deliver the drug product, provides the most accurate representation of patient exposure to leachables by confirming the amount of a substance that has migrated into the drug product. Leachable studies, if designed appropriately, discharge the risk of all failure modes. Nonetheless, leachable studies can reveal high concentrations of organic and inorganic compounds due to unexpected introduction of extraneous chemical species during the manufacturing process and this aspect should be factored in due the probability risk estimation.
Nevertheless, in the absence of leachable data, physicochemical factors relating to the material itself (e.g. porosity, structure, types of extractable species, and etc.), its environment during use (e.g. temperature, type and duration of contact, and etc.) and its extractable profile can be considered to qualitatively assess risk probability (see also Appendix 2). For a more quantitative treatment, mathematical (mass transport) modeling can represent an expedient means to estimate leachables levels (see also Appendix 3). Note that both qualitative or quantitative projection of the impact of physicochemical factors on the accumulation of leachables can complement and/or support experimental data, in particular with respect to the correlation of extractables and leachables.
Extractables data information
An extractable study aims to predict the potential leachable burden from one specific material, components or container closure system. It relies on selecting a solvent system that 1) brackets the different products/formulations that interact with the material, 2) is representative of the commercial drug product, and 3) uses extraction conditions that model manufacture and storage of the product. While the choice of extraction solvents should be guided by best mimicking the propensities of a contacting pharmaceutical fluid matrix, this study is best suited to drug product formulations that are simplistic and easily represented as an extraction solvent, or for materials that will encounter a variety of drug product formulations, e.g., empty pre-filled syringes, IV bags, administration sets. While these studies might be fairly straightforward for sterile Water for Injection (WFI) products, they might be more challenging for complex biopharmaceutical formulations. In these cases, appropriate mixtures of organic and aqueous solvents might be required. These studies are a reliable alternative to leachable studies providing the study design is representative of the environment the material is used in for the commercial product.
Appendix 2. Physicochemical factors impacting the levels and types of leachables in a pharmaceutical preparation (drug solution)
The accumulation of leachables in a pharmaceutical preparation and similarly the sorption of compounds to a plastic material are driven by (i) thermodynamic (i.e., partitioning, solubility) and (ii) kinetic constraints (i.e., diffusion, material diffusivities). A number of physicochemical factors imposed by the system and its constituents with a potential to migrate between the system phases impact both partitioning and diffusion behaviour and are specified below.
A2.1 Thermodynamic factors: partitioning and solubility limit
Partitioning of a compound between a (plastic) material and a contacting pharmaceutical preparation representing two homogeneous phases relates to its differential solubilities in both phases. A compound’s partition coefficient is a key determinant dictating its maximum level of accumulation in the pharmaceutical preparation. This is discussed further in Appendix 3 on Mathematical Modeling (Mass Transport Modeling), eq. (1) and (2).
Limiting solubilities constrain the maximum accumulation of leachables to their level of saturation in the contacting solution (liquid drug product or process stream). They are thereby important physical determinants impacting probability of occurrence (see also Figure A3.1).
A2.2 Solubilization strength of the pharmaceutical preparation (drug solution)
The solubility of an extractable compound in the pharmaceutical preparation depends on its composition. While polar (hydrophilic) compound show good solubility or even miscibility in purely aqueous solutions the solubility of more hydrophobic compounds strongly increases in the presence of organic solubilizers (for example, surface-active agents (polysorbate 80) or co-solvents (e.g., ethanol)) thus increasing the propensity of the preparation to extract/accumulate constituents from the contacting material. Some materials (e.g., polyamide) are known to swell upon solvent contact and this should be taken into consideration when assessing the probability of the risk.
A2.3 Clearance and fate of leachables
During manufacturing of biopharmaceuticals, there is potential to reduce the likelihood of the risk by clearance of leachables during steps including ultrafiltration, diafiltration, chromatography or others. Such processes can effectively dilute or even reduce (purging, fate of leachables) leachables levels thus potentially reducing the probability of a failure mode occurring.
A2.4 Factors affecting migration kinetics (time-to-equilibrium)
Material structure and morphology
The physical characteristics imposed by structure and morphology of materials of construction can play a role in analyzing the probability of the risk as packaging materials display different porosities, water barrier properties, glass transition temperature, densities, etc. While these materials characteristics impose primarily compound diffusion but also partitioning, they can be considered when performing a risk analysis.
Sterilization technology
Sterilization of closure container systems can be done by steam, but other technologies are also employed for molded parts (e.g., ethylene oxide, gamma-irradiation). The effect of these different techniques can affect the probability of leachables to migrate into drug solution by alteration of the material’s physical properties. Additionally, by introducing energy or reactive chemicals (e. g. ethylene oxide) into the material, sterilization processes might contribute to degradation-/reaction products altering the profile of compounds with a potential to leach.
Temperature
Diffusion / migration processes are distinctly accelerated as a result of a temperature increase. Therefore, when systems are exposed to elevated temperatures during processing, storage and distribution, this typically contributes to higher leachables levels after a certain time-period, shorter time-to-equilibrium and therefore a generally increased risk of leaching to occur, respectively.
Contact time
The probability of a leachable to cause harm increases with longer exposure times of the materials in contact with solution due to dependency of compound migration on time (see also eq. (3), Appendix 3). This is applicable, but not limited to:
container closure/bag systems in contact with liquid and/or solid drug ingredients/formulations
time of residence or contact in biopharmaceutical manufacturing systems (vessels, tubing, connectors, filters etc.)
sterilization time
Contacting material surface area to volume ratio
The ratio of the material’s contacting surface area towards the pharmaceutical preparation and the material’s own volume (SA/Vmat) is decisive for the kinetics of migration as it represents the ratio of the cross section leachables need to pass in relation to the total pool of leachables in the material and thereby available for mass transport. A high surface area-to-volume ratio promotes the rate of mass transport (leaching) and shortens the time for equilibration of the system. This results in an overall greater likelihood for substances to migrate from the material.
This ratio should not be confused with the material’s contacting surface area towards the pharmaceutical preparation and the volume of the pharmaceutical preparation itself (SA/VPhP) as the latter is only deemed to indirectly affect partitioning.[20]
Appendix 3. Mathematical modeling (mass transport modeling)


Risk Review and Lifecycle
Appendix – Risk Identification and Risk Statements
As noted, risk assessment may be done in a number of different ways. One approach is FMEA. Within an FMEA framework risk identification can be described and documented as risk statements. These are typically structured as:
Because of………. (the cause) there is a risk that………. (risk event) resulting in………. (the effect).
Well-written risk statements relevant to change management can ensure that risks related to change are well understood by a diverse team of SMEs and thus can be assessed effectively and efficiently (Table A.1).
