
Risk Control-Knowledge Gathering
Extractables and Leachables Management within a Risk-based Framework – Knowledge Gathering for Chemical Risk Control

Introduction
Management of leachables is an important aspect of ensuring the quality and safety of many small and large molecule drug products. The scope of this management generally entails information gathering, risk assessment, and extractables evaluation on materials of construction, packaging components, drug delivery devices and/or their components, and/or components used in the manufacturing process (e.g., single use system components), and potentially leachables studies on final product. Ultimately, leachables management should be done within a rationalized risk-based framework, i.e., a risk management process that provides the pharmaceutical manufacturer with the means to determine the most impactful, meaningful testing and information gathering strategies to ensure drug product quality and patient safety. A key part of the pharmaceutical risk management process is what may be termed, “risk control” – actions taken to reduce risk to an acceptable level for a given product. In this section, we will consider what risk control might mean with respect to gathering chemical information and data relevant to leachables management. We will note helpful resources that already exist in describing this aspect of risk control for leachables. We will also note gaps and inconsistencies that exist in current regulatory documents on this topic and which should be addressed or harmonized to advance quality, safety, and more efficient product development. Another major aspect of risk control is toxicological risk assessment.
Background
Risk control is part of a larger risk management process. An excellent high-level conceptual framework and description of quality risk management is provided by the ICH Q9 guideline.[1] Key aspects of this framework are noted in the main Introduction.
This general process can be used effectively to manage leachables. As described in ICH Q9, risk control activities may include risk reduction and actions to mitigate and accept risk when it exceeds a specified level.
Information and gathering of prior knowledge during materials selection from the supplier or historical data generated in house on similar products are key factors in the risk assessment. Certificates of compliance to USP and Ph.Eur. monographs along with certificate of analysis that the raw material doesn’t include hazardous material are critical for assessing the material from an E&L standpoint (see materials selection).
In case this knowledge is inadequate or insufficient to establish low risk or has a high hazard appraisal (based on the hazard appraisal matrix (see materials selection) activities will include gathering additional information to allow the pharmaceutical manufacturer to make a decision on how to reduce the risk and whether to accept the risk. Generally, this information aims to reduce specific risks identified during risk assessment.
Figure 1 provides examples of (i) types of information that can be used to reduce risk; and (ii) an estimate of their relative ability to reduce risk. The chart is not meant to convey absolute values or ranking for each effect; relative effect may be impacted by the relevance and quality of the data.
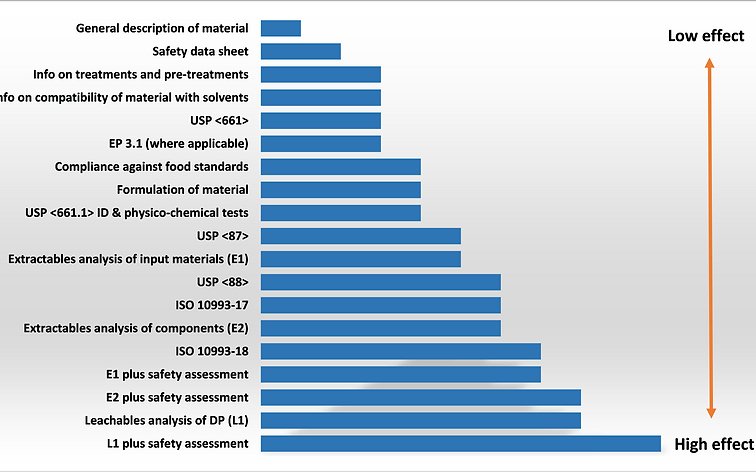
Figure 1. Example of information types able to affect risk control and their relative potential effect. E1 = Extractables analysis of input materials; E2 = Extractables analysis of components; L1 = Leachables analysis of drug product (DP). As noted below, extractables and leachables studies can be done according to pharmacopoeial or best practices resources.
As illustrated, the information obtained can play a role in risk reduction. For example, if a material is considered to represent an unacceptable risk to drug product safety or quality, it might be possible to replace it with another material of lower risk, e.g., a material that does not contain substances of concern or is known to leach less. Further, it might be possible to reduce leachables risk by coating a material to prevent substances leaching or slightly modifying the manufacturing process (e.g., introducing pre-washing or purging steps). Finally, analytical studies can reduce risk by measurement of actual patient exposure followed by a safety risk assessment of results, rather than an assumption that substance present in the material do leach.
Documents such as the PQRI recommendations for extractables and leachables in orally inhaled and nasal drug products (OINDP); the EMA guideline on plastic immediate packaging; the BPOG extractable study protocol and leachables guideline; the USP <661> series, draft <665>, and chapters <1663>, <<1664>, and draft <1665>; among others, are examples of relevant and helpful documents that provide specific information on risk control activities, with respect to leachables management.[2], [3], [4], [5], [6], [7], [8], [9], [10], [11], [12]
Further, different types of testing and/or information gathering can be applied to various levels of the supply chain (e.g., raw material supplier, converter, manufacturer) for different component types such as plastic, elastomer, metal/glass, multi-layer materials (e.g., foil). The IPAC-RS materials quality baseline requirements document provides some helpful perspectives on this risk-based, tiered approach. The IPAC-RS document as well as ICH Q3D on elemental impurities, and others, notes that again, based on risk assessment, information relevant to sources of elemental impurities as potential leachables may be helpful to obtain from the supply chain. [13], [14] [15]
The ability to share such information across the supply chain will rely on strong partnerships among suppliers and the final product manufacturer, enhanced and formalized through quality and technical agreements that may define the types of testing and information that could be shared, and agreed access to drug master files.
GAP: Although there are a number of informative and generally accepted publications including industry practices and compendial chapters that describe how to design and implement risk control activities, e.g., extraction studies, leachables studies, there is no overarching harmonized regulatory guidance that positions these documents and studies within a holistic risk management framework, and which also provides insights on the hierarchies of information that such studies provide.
Extractable Study Design Concepts
Extractables are organic and inorganic chemical entities that are extracted from a material or component under laboratory conditions using the umbrella term: extractable studies. As such, they are designed experiments and the choices made in the design can profoundly affect the results obtained. These have been given a variety of names that reflect different design choice made to attempt to accelerate, exaggerated or simulate the process of leaching. They also have been linked to purpose descriptors such as scouting, discovery, identification and quantification.
Design considerations include:
Extraction type
Extraction solvent
Extraction temperature and/or pressure
Extraction time
Preparation method from material (cutting, milling, flushing etc.)
Surface area of material or component to extraction solvent volume ratio
The general purpose of conducting an extractable study is to generate an extractables profile, with information on identity and quantity of observed extractables, from a material covering the wide range of extractables using a number of orthogonal analytical methods capable of detecting of a wide range of different extractables compounds classes (e.g., volatiles, semi-volatiles, and non-volatile extractables including elemental impurities).
Within a risk management framework, extraction studies should be designed to link to specific areas of risk identified in the risk assessment. For example, within an FMEA approach, the study may be designed to link to a failure mode(s), such as, “Because of the interaction between the formulation and the elastomer valve seal, there is a risk that leachables from the seal enters the formulation and are dosed to the patient resulting in harmful exposure.” As a part of risk control data gathering to impact decision making, extraction study design generally reflects a particular intent related to the types of data needed for the risk control activity.
For example, extraction studies may be designed to guide development of leachables methods (method development) and/or designed to investigate potential leachables, where solvent selection and extraction techniques may be broad and fairly comprehensive. The extraction study might also be designed to simulate drug product leaching conditions, potentially as a substitute for leachables studies (so called “simulation studies”). These are all extraction studies designed and implemented to serve specific purposes within a risk control milieu (Figure 2).
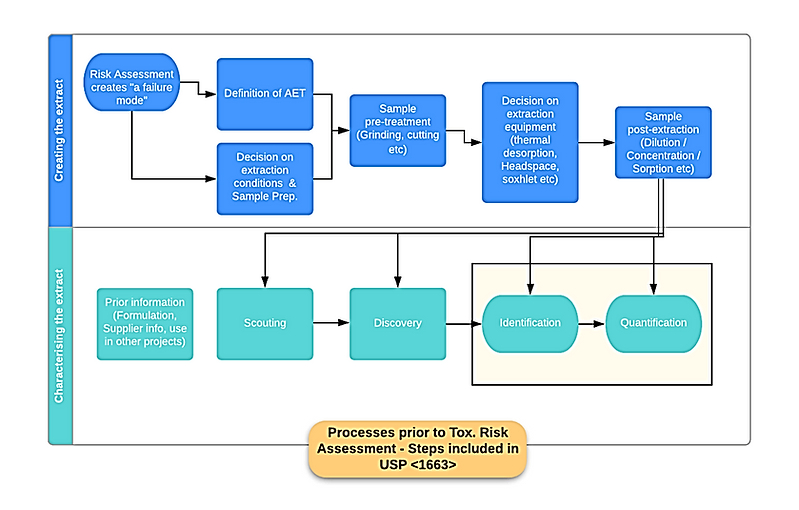
Figure 2. Example process steps for extractable studies for risk control, from USP <1663>
As noted in the introduction, current key sources that provide specific considerations on extractable study design include USP <1663>, which provides guidance for the design and execution of extractables assessment for pharmaceutical packaging and delivery systems. For polymeric single-use systems used in biopharmaceutical manufacturing, the BioPhorum Operations Group (BPOG) has provided an analytical protocol for suppliers and others to use when measuring extractables in these materials.
Simulation Studies
One type of valuable and informative study design is the “simulation study.” This type of study as well as considerations for when such studies could be used and justified are described in various pharmacopoeial chapters and industry case studies.[7], [8], [16]
The aim of these studies is to closely match to outputs that would have been obtained if leachable studies were conducted, acting as a substitute in situations where leachable studies are difficult to complete. There are a number of reasons why a leachables study may be difficult to complete, and these may include:
Complex drug product formulation matrices which restrict accurate leachable determination (creams, ointments, presence of interfering excipients or highly concentrated protein solutions, etc.)
Leachables samples are not suitable (e.g., samples are from early development or formulations of cell-based or biological samples)
The challenge in prediction of potential leachables in a drug product based on extractables testing is that extractables study will use extraction solvents and that the leachables study will use the drug product formulation as solvent. The polarity, pH and ion strength of the extraction solvents will differ from that of the drug product formulation. Leaching into a soluble drug product is a function of polarity, pH and ion strength, often referred to as “like dissolves like”, meaning that leachables will leach into a compartment where the polarity, pH and ion strength is as close to the leachables own properties for those factors. The difference is often referred to as the gap between extractables and leachables testing and are especially pronounced for highly complex drug product formulations such as biopharmaceuticals typically containing a protein in a high concentration which is blended in with an isotonic agent, a preservative, a buffer, a surfactant. Consequently, causing the polarity, pH and ion strength to be quite different from that of the extraction solvents.
A well-designed simulation study has the potential to close the gap between extractables and leachables and predict actual leachables that will be observed in the drug product. If the simulation study can use the drug product formulation as solvent it can furthermore be used as a risk mitigation tool in early development to investigate potential chemical interaction/reaction between leachables and the active pharmaceutical ingredient (API) as well as with formulation components. Such interactions, which are highly likely to occur between leachables and proteins, can affect safety and drug product quality.[16]
GAP: Currently it is not clear whether or how regulatory agencies are accepting simulation in lieu of leachables studies. Consideration of such studies within harmonized regulatory guidance, particularly within the context of a risk management framework would be very helpful to industry.
GAP: There is a need to clarify health authorities’ views on the impact of proteins as well as lipids and other excipients on leaching.
Leachable Study Design
Leachables studies can provide highly impactful information regarding understanding which leachables will be present in final drug product, and to what patients will be actually exposed to. Data from leachables studies, along with a safety assessment of leachables, can provide key practical information that allows users to mitigate risk and/or make decision regarding risk acceptability. Within a risk management framework, such information could be very impactful in reducing probability scores in an FMEA model. Simulated studies could also have similar impact and importance depending on the product profile and the risk assessment (see above for further details).
The leachables study is an analytical investigation to identify qualitatively and quantitatively, leachable(s) that patients are exposed to over the proposed shelf-life of a particular drug product from the packaging or delivery system, and production systems. Leachables studies are required to support a comprehensive safety evaluation of a drug product mainly when the degree of concern associated with the route of administration is high and/or there is high likelihood of interaction between the packaging component or dosage form with the drug product. Leachables studies can be used to establish the specifications and drug product acceptance criteria for leachables that pose risks related to patient safety or drug product quality. Leachables studies can be executed at any stage of product development, although mainly at clinical phase III, and prior to file submission on the final formulation and packaging system. There are a number of key documents that provide information on the conduct and purpose of leachables studies. [2], [3], [4], [8]
The design of a leachables study is case dependent as it relies on the particular purpose of assessing a certain drug product and requires gathering key information such as the extraction profile, chemical composition of the material of construction of the packaging/delivery system and details of the manufacturing process and clinical use. [17]
Thus, the leachables study design for the packaging or delivery system should consider the following:
The assessment should be executed on drug product formulation (lyophilized powder, liquid, suspensions, etc.) with final packaging and delivery system including both primary (glass or plastic) and secondary packaging (adhesive labels, printed over pouch, aluminium seal, etc.)
Experimental conditions should reflect the actual manufacturing and storage conditions for, e.g., sterilization program, storage time and temperature, clinical use, etc.
Auxiliary systems involved in the product clinical administration (e.g., infusion bags, transfer sets) should also be considered in the study design, if appropriate.
Accelerated storage can be performed depending on the nature of the drug product, and data can be provided at least during submission, along with leachables data from long-term storage conditions.
Per stability testing, three replicates are required per assessment on multiple formulation batches and one batch of the packaging system that can be tested simultaneously, although there are important questions regarding this practice (see “Gaps” below).
If a product comprises a range of formulation strengths, a bracketing approach or selection of a worst-case set of conditions can be applied to cover all formulations.
The leachables study design for production systems should consider the following:
The assessment should be executed on components as used during production (e.g. sterilized assemblies or individual components) with the production step process stream (i.e. specific buffer/solvent system, formulated bulk drug substance, or drug product formulation).
Experimental conditions should reflect the actual manufacturing conditions for, e.g., sterilization program, contact time, process temperature, static vs. dynamic exposure, etc.
Sampling directly from the production floor provides the most relevant samples but may be accompanied by other challenges(e.g., cannot isolate a specific component, risk to sterility of the process, available production times not aligned with testing needs, risk of contamination)
In particular, simulation studies may be more appropriate and achievable from a technical and practical point of view for production systems (due to complex formulations, provides opportunity to isolate each component to trace source of leachables, timing is not dependent upon production of a batch, etc.)
For components utilized in multiple production steps, leachables testing for the highest risk step can be used to support the use in all steps.
Safety assessment should include the leachables from overall, cumulative use of a specific component.
The risk assessment process can be a useful guide to the design of the leachable study highlighting the focus areas and providing a justification for nature of the protocol established.
GAP: Currently, stability testing approaches are applied to leachables testing, i.e., leachables information is obtained using three lots of drug product (DP) batches. However, for leachables testing on the container closure system (CCS) the use of three DP batches holds no value for information related to variation of leachables identities and concentrations from the CCS. Consideration of leachables variation form CCS within harmonized regulatory guidance, particularly within the context of a risk management framework would be very helpful to industry.
GAP: Management of auxiliaries for drug products clinical administration such as catheters, infusion bags, transfer sets, etc. is unclear with respect to responsibilities and requirements in terms of E&L. What is expected needs to be clarified, considering the short contact of the solution with the material, but on the other hand the direct and immediate contact with the patient.
Barrier to Risk Reduction within Extractable and Leachable Studies
Analytical Method Thresholds
Creating analytical methods which are fit for purpose is a key aspect of E&L studies. Key components include detection, identification and quantification thresholds.
Such thresholds provide a rationalized means to assess the presence of extractables and leachables and collect meaningful data on them. The application of such thresholds allows for risk reduction and can reflect the level of risk a pharmaceutical manufacturer is deciding to accept. Analytical method thresholds are thus a useful part of risk control strategies. Such thresholds help establish the operating range and threshold level at which an analytical method is expected to perform to accomplish its appropriate functions and can be used by suppliers and final drug product manufacturers in extractables and leachables studies. As noted in USP <1664>[8], an appropriate method must function at levels greater than or equal to the analytical threshold.
These thresholds can be based on generic safety thresholds or individual compound permitted exposure levels when tied to safety risk assessment but could also be study specific.
The Analytical Evaluation Threshold (AET) concept created and proposed in the PQRI OINDP recommendations is an example of a safety-derived threshold. An AET is defined as “the threshold at or above which, a particular extractable and/or leachable should be identified, quantified and reported for potential toxicological assessment.” Application of the AET concept is well-described in the PQRI recommendations as well as the USP <1664>. An AET can also be derived using other scientifically based safety threshold values (see risk control – safety assessment).[18]
In the case of elemental impurities, analytical threshold limits can be calculated based on permitted daily exposure limits provided in the ICH Q3D for the most toxic elements.
A few considerations regarding analytical method thresholds and method capability are provided as follows:
Analytical method performance (i.e., instrumental sensitivity, analytes recovery) might not be immediately adequate to meet the analytical threshold if it is too low (based on product dosage). In such cases, different techniques can be evaluated; furthermore, the development of appropriate sample preparation can be used to improve method sensitivity.
Relative response factors are also important aspects to consider especially for screening/untargeted methods where the semi-quantification is carried out using selected reference compounds (internal/external) to quantify classes of compounds with similar structures and/or physico-chemical characteristics. Similarly, the levels of unidentified compounds (unknowns) or species for which authentic reference compounds are not available can be estimated using selected surrogate reference compounds. The relative response factor of unknown species with respect to the reference compounds might be considered a gap here. This uncertainty in the estimation can be overcome by applying a safety margin to be sure to not underestimate the real levels.
The Influence of Degree of Certainty Ascribed to an Extractable or Leachable
Detection and then identification of either extractables or leachables above an analytical method threshold is an integral part of the risk control knowledge gathering process as identification and quantification is important to safety risk assessment cannot proceed without a quantified amount and established identity. If identification is uncertain, then risk control either cannot move forward or will proceed with potentially false assumptions.
Generally, identification categories ascribed to substances are: Confirmed, Confident, Tentative and Unknown [2], [7] and alternative terms, e.g. “most probably,” should be discouraged. Structural information and the level of confidence in identification of the extractable or leachable are essential for a meaningful toxicological risk assessment to assess patient safety and mitigate the risk. Substances described as Unknown or Tentative are unlikely to be useful in risk control.
Typically, the mass spectrum of a substance is matched against a spectral library to identify the extractable or leachable. However, care should be taken to avoid misidentification. Spectral libraries contain a limited number of substances, and false positives can occur when the spectra are matched against the wrong candidate. The best hit from a spectral library is often reported as the most likely candidate, however alternative hits should be considered. A high-quality spectrum is important for a good spectral library match. Data is generally processed to obtain a pure spectrum and discard artefacts or separate co-eluting peak. If too much information is discarded this can result in false negatives. Spectral libraries are mass analyzer specific, and the spectrum of a compounds depends of the ionization technique and applied voltage.
The USP <1663> provides a basic framework for criteria to apply and this is built upon in a paper by Booij and Creasey.[19] Both of these confirm a reference standard is essential for confirmation of the identity of a substance supported by spectroscopic data. The accurate mass, retention time match on the same chromatographic system and other spectroscopic information (e.g., fragmentation data, collision cross section) of the reference material should be compared to the substance of interest to confirm the identity. However, a reference material is not always available in situations such as this. A structure can be supported with a well thought out approach, e.g., good spectral library match, characteristic fragment ions or molecular ion, accurate mass and retention time and includes where necessary data from orthogonal techniques (e.g., NMR, FTIR). Alternatively, a reference material can be synthesized to confirm the identity of a substance. These approaches require additional analytical techniques, equipment and expertise, which can be a barrier and limit the identification and confirmation of extractables and leachables. There is a need for practical guidance and approaches for data processing using different techniques and confirmation of substances.
GAP: Health authority guidelines currently do not address the risk associated with the difference in response factors for unknown species when their concentration or amount are estimated using reference compounds with another identity. Further, the industry needs practical guidance and approaches for data processing using different techniques and confirmation of substances. Harmonized regulatory guidance that includes concepts regarding how to handle differences in analytical response factors, approaches for data processing using different techniques, and confirmation of substances, particularly within the context of a risk management framework would be very helpful to industry.
Progression to Risk Review
Risk control activities may need to be re-assessed or performed during risk review activities including lifecycle management, e.g., change management. Risk control may also lead to further risk assessment activities (e.g., aspects of FMEA, risk ranking, etc.) in situations where, for example, risk reduction activities might introduce new risks.
References
[1] ICH Q9, Quality Risk Management. International Conference on Harmonisation of Technical Requirements for Registation of Pharmaceutials for Human Use. Step 4, November 2005.
[2] Safety thresholds and best practices for extractables and leachables in orally inhaled and nasal drug products. Product Quality Research Institute. September 2006.
[3] Guideline on Plastic Immediate Packaging Materials. European Medicines Agency. December 2005.
[4] Best practices guide for evaluating leachables risk from polymeric single-use systems used in biopharmaceutical manufacturing. BioPhorum Operations Group.
[5] Ding W, Madsen G, et al., Standardized Extractables Testing protocol for single-use systems in biomanufacturing. Pharmaceutical Engineering, Vol. 34, No. 6. November/December 2014.
[6] USP <661>, <661.1>, <661.2> Plastic Packaging Systems and Their Materials of Construction; Plastic Materials of Construction; Plastic Packaging Systems for Pharmaceutical Use. United States Pharmacopoiea.
[7] USP <1663> Assessment of Extractables Associated with Pharmaceutical Packaging/Delivery Systems. United States Pharmacopoiea.
[8] <1664> Assessment of Drug Product Leachables Associated with Pharmaceutical Packaging/Delivery systems. United States Pharmacopoiea.
[9] USP <665> Plastic Materials, Components, and Systems Used in the Manufacturing of Pharmaceutical Drug Products and Biopharmaceutical Drug Substances and Products.
[10] USP <1665> Characterization of Plastic Materials, Components, and Systems Used in the Manufacturing of Pharmaceutical Drug Products and Biopharmaceutical Drug Substances and Products.
[11] D. Jenke, J. Castner, T. Egert, T Feinberg, A. Hendricker, C. Houston, D.G. Hunt, M. Lynch, A. Shaw, K. Nicholas, D.L. Norwood, D. Paskiet, M. Ruberto, E.J. Smith, F. Holcomb. Extractables characterization of five materials of construction representative of packaging systems used for parenteral and ophthalmic drug products. PDA J Pharm Sci Technol. 76(5): 448-511 (2013).
[12] FDA Guidance for Industry. Container Closure Systems for Packaging Human Drugs and Biologics. United States Food and Drug Administration, May 1999.
[13] Baseline Requirements for Materials Used in Orally Inhaled and Nasal Drug Products (OINDP). International Pharmaceutical Aerosol Consortium on Regulation and Science. February 2017. https://ipacrs.org/news-events/news/ipac-rs-updates-recommended-baseline-requirements-for-materials-used-in-oin
[14] Jenke D. Materials in Manufacturing and Packaging Systems as Sources of Elemental Impurities in Packaged Drug Products: An Updated Literature Review. PDA Journal of Pharmaceutical Science and Technology, 2019.
[15] ICH Q3D(R1). Guideline for Elemental Impurities, March 2019.
[16] Li K, Rogers G, Nashed-Samuel Y, Lee H, Mire-Sluis A, Cherney B, Forster R, Yeh P, Markovic I. Creating a Holistic Extractables and Leachables (E&L) Program for Biotechnology Products. PDA J Pharm Sci Technol.; 69(5):590-619 Sep-Oct 2015.
[17] Guidance for Industry Q1A(R2) Stability Testing of New Drug Substances and Products.
[18] ICH M7(R1). Assessment and Control of DNA Reactive (Mutagenic) Impurities in Pharmaceuticals to Limit Potential Carcinogenic Risk. Step 4. March 2017
[19] Booij, P, Creasey, JM. Identification of unknown extractables and leachables using mass spectrometry: Identification with confidence? BioPharma Asia, Vol. 8, Issue 6 November/December 2019.